MyPro includes the following software in a Virtual Box, please make sure
you have accepted their license agreements if any.
|
Name |
Download |
Reference |
Checkbox |
|
Abyss 1.5.2 |
http://www.bcgsc.ca/platform/bioinfo/software/abyss/releases/1.5.2 |
(Simpson, et al., 2009) |
□ |
|
Bio-Linux 8 |
http://environmentalomics.org/bio-linux-download/ |
(Field, et al., 2006) |
□ |
|
CISA 1.3 |
http://sb.nhri.org.tw/CISA |
(Lin and Liao, 2013) |
□ |
|
Edena V3.131028 |
http://www.genomic.ch/edena.php |
(Hernandez, et al., 2008) |
□ |
|
FastQC 0.10.1 |
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
|
□ |
|
Prokka 1.10 |
http://www.vicbioinformatics.com/software.prokka.shtml |
(Seemann, 2014) |
□ |
|
r2cat |
http://bibiserv2.cebitec.uni-bielefeld.de/cgcat?viewType=download |
(Husemann and Stoye, 2010) |
□ |
|
SOAP2 2.21 |
http://soap.genomics.org.cn/soapaligner.html |
(Li, et al., 2009) |
□ |
|
SOAPdenovo 2.04 |
http://sourceforge.net/projects/soapdenovo2/files/SOAPdenovo2/bin/ |
(Luo, et al., 2012) |
□ |
|
SPAdes 3.1.1 |
http://bioinf.spbau.ru/spades |
(Bankevich, et al., 2012) |
□ |
|
Tablet 1.14.04.10 |
http://ics.hutton.ac.uk/tablet/download-tablet/ |
(Milne, et al., 2013) |
□ |
|
VelvetOptimiser 2.2.5 |
http://bioinformatics.net.au/software.velvetoptimiser.shtml |
(Zerbino and Birney, 2008) |
□ |
Please
note that Abyss was made with maxk=128, Velvet was
made with MAXKERLENGTH=127.
Installation of MyPro
Download Virtual Box
https://www.virtualbox.org/wiki/Downloads
Download MyPro.ova:
To
download http://sb.nhri.org.tw/MyPro.ova
(5.5GB) or http://sb.nhri.org.tw/MyPro.tar.gz
(5.4GB)
Other
information at: http://sourceforge.net/projects/sb2nhri/files/MyPro/
Open VirtualBox
File
-> Import Appliance...
Select
the file (MyPro.ova) to import
MyPro.ova
Or,
directly double click on MyPro.ova
Please
check the box of "Reinitialize the MAC address of all network cards"
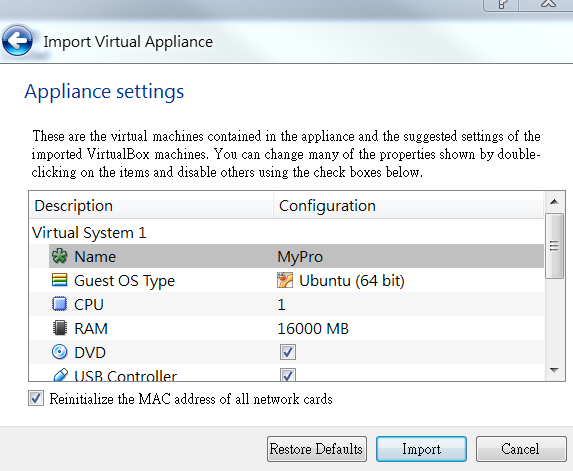
Import
Open Terminal
Quick-start
guide of MyPro
A. Pre-process
This
script is used to trim, pair and sub-sample your raw
reads. A total of 100X (paired) reads are generated. This process is strongly
recommended; otherwise much computational time is required for genome assembly.
Command:
Preprocess.py
-read1 S.gordonii_G9B_TTAGGC_L001_R1_001.fastq -read2
S.gordonii_G9B_TTAGGC_L001_R2_001.fastq -g 2200000
Preprocess.py -h
for help.
B. AutoRun
This
script is used to perform Assemble, Integrate and Annotate.
Command:
AutoRun.py G9B -read1 50X_R1.fastq -read2 50X_R2.fastq -evaluate
-p '--prefix G9B --genus Streptococcus --species gordonii
--strain G9B --addgenes --locustag
AA01'
AutoRun.py -h, Assemble.py -h, Integrate.py -h
and Annotate.py -h for help.
C. Post-assemble
This
script is to (1) merge your ordered contigs if they
are overlapped and (2) fill gaps with the contigs of
local assembling.
(Optional)
To use r2cat.jar for ordering your contigs
against a related reference genome. You can Export contigs
order (FASTA) and Export unmatched contigs (FASTA)
separately.
Command:
Postassemble.py
-o cisa.ordered.fa -u unmatched.fa
-read1 ../50X_R1.fastq
-read2 ../50X_R2.fastq
No
reference genome:
Postassemble.py
-u cisa.ctg.fa -read1 ../50X_R1.fastq -read2 ../50X_R2.fastq
Postassemble.py -h for help.
MyPro has been published in Journal of Microbiologic Methods (http://www.sciencedirect.com/science/article/pii/S0167701215001207). Please download http://sourceforge.net/projects/sb2nhri/files/MyPro/Supplementary%20Information.pdf/download for details.