The World Health Organization (WHO) organizes consultations in February and September of each year, spearheaded by an advisory group of experts to analyze influenza surveillance data generated by the WHO Global Influenza Surveillance and Response System (
GISRS). The purpose of these consultations is to recommend the composition on influenza virus vaccines for the northern and southern hemispheres, respectively. The latest news of influenza viruses is made available to the public and updated on the
WHO website. Although WHO discloses the manner in which it has made the recommendation, usually by considering epidemiological and clinical information to analyze the antigenic and genetic characteristics of seasonal influenza viruses, most individuals do not possess an understanding of antigenic drift and when it occurs.
We have constructed a web server, named Fluctrl, and implemented a pipeline whereby HA sequence data is downloaded from the
Influenza Virus Resource at NCBI along with their isolation information including isolation year and location, which are parsed and managed in MySQL database. By analyzing the frequency of each amino acid residue of the HA1 domain expressed by the viruses on annual basis, users are able to obtain evolutionary dynamics of human influenza viruses corresponding with epidemics. In addition, a distribution of amino acid residues at a particular site is represented geographically to trace the location where antigenic variants are seeded.
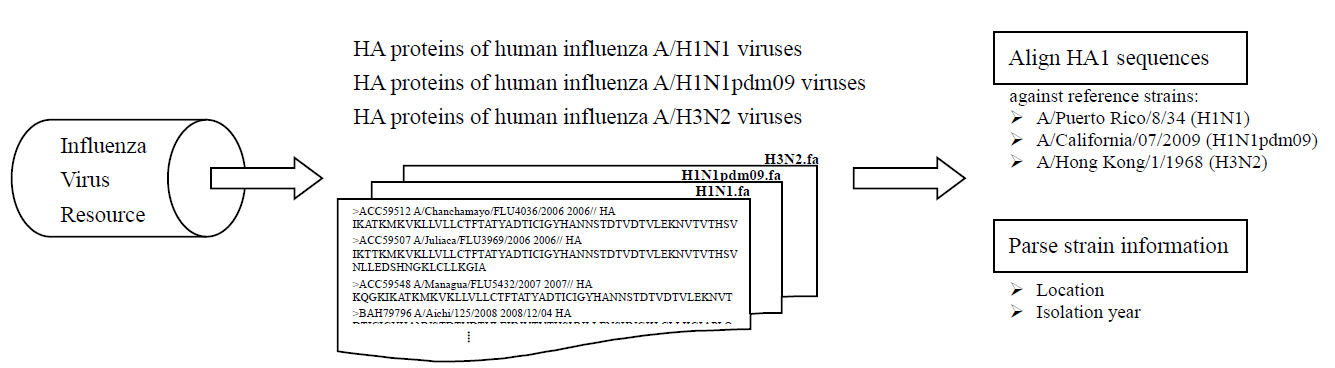
HA protein sequences of human influenza A/H1N1, A/H1N1pdm09 and H3N2 viruses were downloaded separately from the NCBI Influenza Virus Resource (on Jan 06, 2013). For each subtype of human influenza A virus, sequences were aligned against the reference sequences, A/Puerto Rico/8/34 (
YP_163735), A/California/07/2009 (
ACP41953) and A/Hong Kong/1/1968 (
ACC66318), respectively by utilizing
MUSCLE. Currently, 5811 H1N1, 10061 H1N1pdm09 and 12599 H3N2 HA1 viral sequences are available in Fluctrl. Each of the resulting alignments was then transformed to position-wise amino acid residues.
[Latest News on Jan 24, 2017]
Updated HA sequences to the fourth quarter of 2016. Currenlty, 6314 H1N1, 16699 H1N1pdm09 and 24321 H3N2 HA1 viral sequences are available in Fluctrl.
[Latest News on Oct 03, 2014]
Updated HA sequences to the third quarter of 2014. Currenlty, 6314 H1N1, 13701 H1N1pdm09 and 17055 H3N2 HA1 viral sequences are available in Fluctrl.
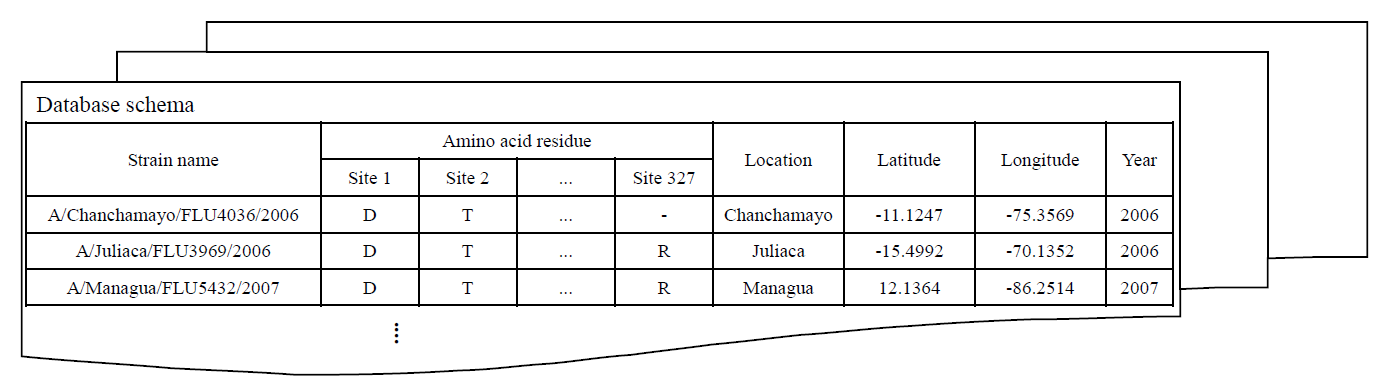
Strain information including location and isolation year were parsed from strain name. Location names were subsequently queried with
Google Geocoding API version 3 to obtain latitude and longitude coordinations. Three SQL tables of H1N1, H1N1pdm09 and H3N2 were created and managed by MySQL.
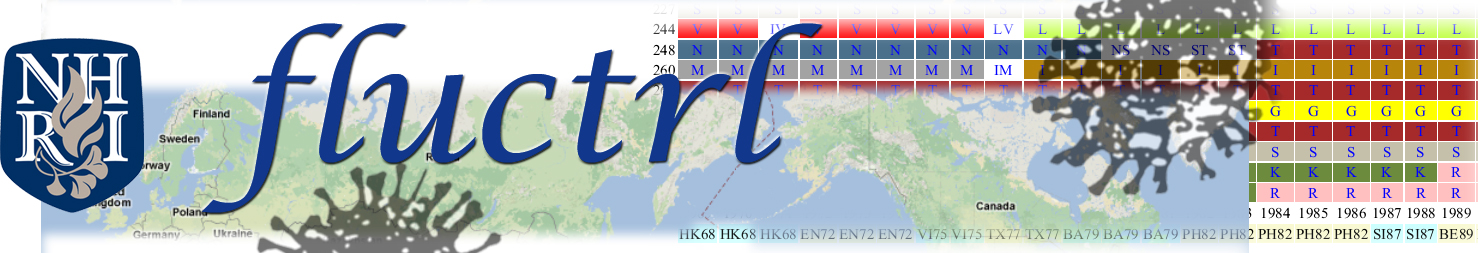
Sequences isolated from the same year were clustered into a single group to obtain the frequency of amino acid residues at each amino acid site. For every amino acid site, if one amino acid residue reached a frequency of >= 0.7 druing a given year, it was assigned as a single "major amino acid residue (MAA)" corresponding to that year (
Liao, YC et al.). Alternatively, multiple MAAs were assigned if more than one residue, whose frequency resided between 0.2 and 0.7, was discovered. The assembled MAAs throughout the isolation years examined represent an evolutionary dynamics perspective of human influenza viruses. The varied and white colors were used to label single and multiple MAAs, respectively. Therefore an evolutionary dynamic pattern of HA1 proteins was clearly demonstrated.
An evolutionary dynamics of human influenza viruses corresponds with epidemics:

For example, the substitutions of K158N, K173Q and N189K could explain the circulating viruses in the years of 2008 and 2009 drifted from BR07-like to PE09-like viruses.
Geographic and temporal information show the original location of antigenic variants:
We have implemented
Google Map API version 3 in Fluctrl to generate a graphical distribution of the influenza viruses within a specified period of years upon the coordinates. A graphical distribution map of human influenza A/H3N2 viruses isolated in the year of 2008 is visualized as below:
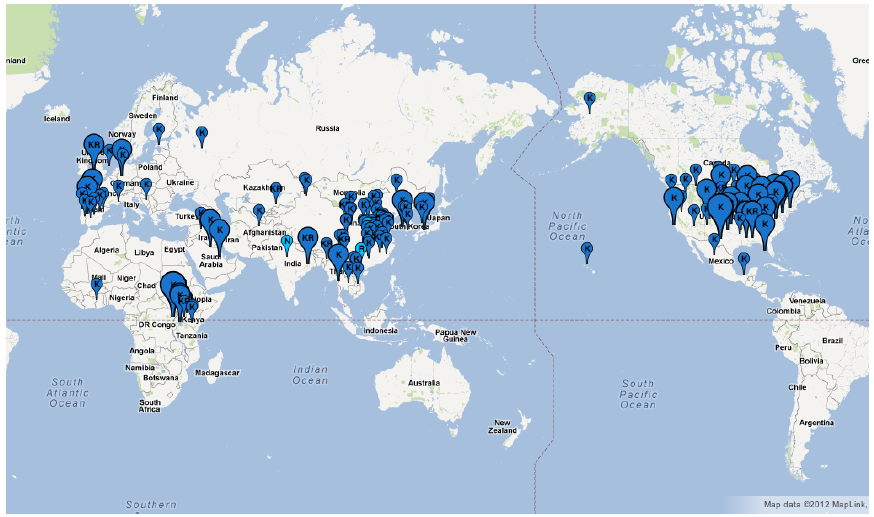
The balloons containing a residue exhibiting maximum frequency at the 158
th site are dark colored; while the other cases are light-colored. The size of the balloons are varied with the size of the sequence: small (No <= 10), medium (10 < No < 50) and large (No > 50).
Another map for human influenza A/H3N2 viruses in 2009:
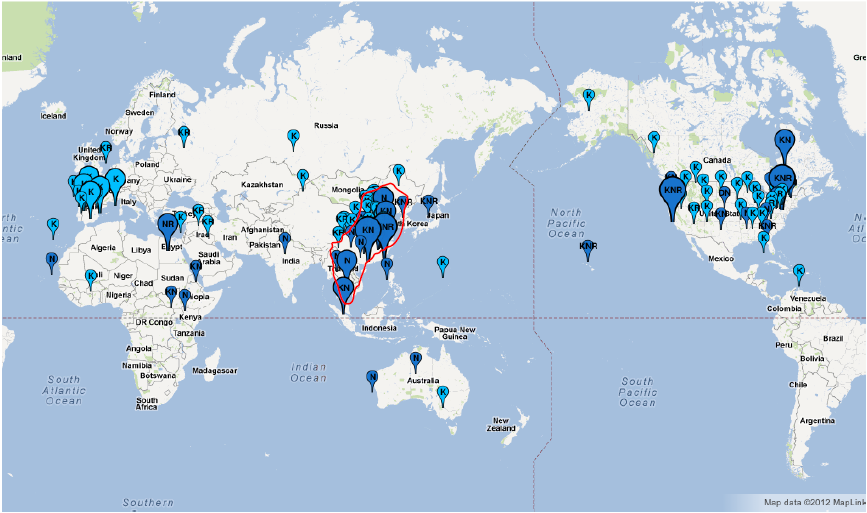
By comparing the 2008 map with the 2009 version, we found that the major amino acid residue on site 158 transformed from K to NK, and those emerging N residues located around East and Southeast Asia, coincided with the fact that new variant influenza viruses emerged from that location [
Yang, JR et al.].
Surveillance within E-SE Asia facilitates vaccine strain selection
Fluctrl provides a functionality for limiting the scope of analyzed human influenza data to E-SE Asia by constraining the latitude (between -8 and 8) and the longitude (between 70 and 150) that were extracted from strain information. Major amino acids (MAAs) in an evolutionary dynamic within E-SE Asia (the right panel) have been observed to change beforehand, compared to the global dynamic (the left panel), e.g. the transitions of L25I,H75Q and H155T are visible in 2002 and the substitutions of A198S and V223I become fixed in 2012. Such differences between the evolutionary dynamic patterns of the world and E-SE Asia imply that surveillance within E-SE Asia would provide accurate monitoring of the emergence of influenza variants when coupled with timely and large-scale HA sequence data.
 Taken together, it becomes practical to detect and monitor the antigenic variants of human influenza viruses with Fluctrl on a continuous basis using the ever-increasing viral dataset to keep abreast of new influenza threats.
Taken together, it becomes practical to detect and monitor the antigenic variants of human influenza viruses with Fluctrl on a continuous basis using the ever-increasing viral dataset to keep abreast of new influenza threats.